�㽭ԭ��ˎeCTD�l(f��)��ܛ�� �\�ŷ���(w��) �x���Ƽ�����(y��ng)
�l(f��)؛���c���㽭ʡ������
�l(f��)���r�g��2025-04-23
�l(f��)؛���c���㽭ʡ������
�l(f��)���r�g��2025-04-23
�W��eCTD�Ěvʷ�ظ��c��(qi��ng)�ƌ�ʩ �W����2003�������M(j��n)eCTD�����ͨ�ü��g(sh��)�ęn���Ę�(bi��o)��(zh��n)���M(j��n)�̣���Ҫ��ˎע����Ո��MAA������CTD��ʽ��2010�꣬���Ќ��u����CP�����ȏ�(qi��ng)��ʹ��eCTD���S���ɢ����DCP���ͻ��J(r��n)����MRP���քe��2015�ꡢ2017����M(j��n)����2019�꣬�W��Ҫ�����Ї��ҳ���NP����ע����Ո����eCTD��ʽ�ύ����(bi��o)־��������f���wϵ�ij��졣2024�꣬EMA����eCTD4.0ԇ�c�Ŀ��ּ���������g(sh��)�������c���uЧ�ʡ� eCTD��C��(bi��o)��(zh��n)�ĵ����c�P(gu��n)�I�� �W�˵���C��(bi��o)��(zh��n)�v��(j��ng)����{(di��o)��������2025��3���õ�eCTD3.1�^(q��)��ģ�����CҎ(gu��)�tv8.1�����ļ��Y(ji��)��(g��u)��Ԫ��(sh��)��(j��)�̓�(n��i)���������������(y��n)���Ҫ��(bi��o)��(zh��n)����ġ�ۙ����Tracking Table������(qi��ng)��У�Ҏ(gu��)�t����15.11��15.12������(d��o)��CEP���W��ˎ���m�����C�����f������ͨ�^���Sռλ�ļ��R�r��Q���c���ڰ汾��ȣ�v8.1��(qi��ng)���ˌ�ģ�Kһ�^(q��)����Ϣ��߉��C������(x��)���ˌ�PDF��������朽ӵ�Ҏ(gu��)���ԙz�顣�Ĵ�����INDע��������P(gu��n)���g(sh��)֧�֡��㽭ԭ��ˎeCTD�l(f��)��ܛ��
���g(sh��)�ډ��c�d�Ј�����(zh��n) ���͖|�ρ������ɼ{eCTD������IT���A(ch��)�O(sh��)ʩ������(d��o)��ʩ�M(j��n)�Ȝ��W��ͨ�^��eCTDȫ���h���ṩ���g(sh��)Ԯ���������d�Ј�������C�wϵ����Ӗ(x��n)���ġ����ˎ����ᘌ���ͬ�^(q��)�����f�����ԣ�������ģ�K1���ӱ��ط�(w��n)���Ԕ�(sh��)��(j��)�� �O(ji��n)�ܿƌW(xu��)�c��(chu��ng)���� eCTD֧���挍�����C��(j��)��RWE�����m��(y��ng)���R��ԇ��O(sh��)Ӌ�����ϣ����ل�(chu��ng)ˎ���С�EMA��PRIMEӋ����ͻ���ԯ����ṩeCTD����ͨ�������S���A���ύģ�K��(sh��)��(j��)����ˎ�̓���ˎ��eCTD���п������M(f��i)�Üp��̓�(y��u)�Ȍ��u�� ����(y��ng)朰�ȫ�c��Ӌۙ eCTD��XML�����ļ�ӛ������ύ�汾��֧�ֹ���(y��ng)朆��}���ݷ�����ԭ��ˎCEP�������輰�r��׃����Ϣ���_�������Ƅ��S�̫@ȡ��(sh��)��(j��)���^(q��)�K朼��g(sh��)ԇ�c����ۙeCTD��(sh��)��(j��)������ֹ�۸ĺ�δ�ڙ�(qu��n)�L���� �Ļ���c��ʩ�ϵK �����ϚW����ƫ�Â��y(t��ng)���|(zh��)���̣���(d��o)��eCTD�ƏV�����^��EMAͨ�^���Z�N��Ӗ(x��n)���Ϻͅ^(q��)��f(xi��)�{(di��o)�T�ƶȴ��M(j��n)�Ļ��m��(y��ng)���ИI(y��)���{(di��o)������˼�S����eCTD�ġ���Ҏ(gu��)ؓ(f��)��(d��n)���D(zhu��n)���顰������(y��u)�ݡ�������CDE eCTD�gӭ�xُ����NDAע��������P(gu��n)���g(sh��)֧�֡�

�^(q��)��c�����f(xi��)������(zh��n) �W��eCTD����ݳɆT���ض�Ҫ������ģ�Kһ��������Ϣ����ϸ����Z�Ժͷ�Ҏ(gu��)������J(r��n)����MRP���У������ɆT����RMS�����u������豻�����ɆT���J(r��n)�ɣ������F(xi��n)��������CMDh�f(xi��)�{(di��o)���ύEMA�ٲá��@�N���Ӽ����u�C(j��)��Ҫ����Ո�����ļ���(zh��n)���A�μ����]�^(q��)������ԣ�������m(x��)�������`�� eCTD4.0��̽���cδ������ ICH��2015��l(f��)����eCTD4.0�汾ּ�ں���Ŀ䛽Y(ji��)��(g��u)��֧�ֶ�a(ch��n)Ʒ��ͣ����t(y��)����е���������(qi��ng)�������ڹ������ܡ��W��Ӌ��ͨ�^2024��ԇ�c���^����4.0�����ƽ���ļ��M����ʽ�����p���؏�(f��)�ύ���������uЧ�ʡ�Ȼ������ʩ���Q�F(xi��n)��ϵ�y(t��ng)�����Լ��ИI(y��)�m��(y��ng)�Ԇ��}��
�������u�����ceCTD�f��;�����m�䣺�W��ˎƷ���u����������У�CP������ɢ��DCP�������J(r��n)��MRP���͇��ҳ���NP����eCTD���m�䲻ͬ������f��Ҫ�����磺 ���Ќ��u����CP����ͨ�^EMA��eSubmission Gateway�ύ�����u�r�s240�������գ�eCTD�����������ģ�K1-5�����Z�Ԙ�(bi��o)���ļ��� ��ɢ���u����DCP������ͨ�^CESP���W�˹�ͬ�ύ�T�����f���������ɆT����RMS������(d��o)���u��eCTD��֧�ֶ���ͬ���u����ģ�K����֡� ���J(r��n)����MRP�������ڙ�(qu��n)�ɆT������RMS��eCTD������������У�Baseline Sequence 0000�������Ϛvʷ���u��(sh��)��(j��)����ͨ�^CMDh�f(xi��)�{(di��o)���硣����API��DMF������P(gu��n)���g(sh��)֧�֡�
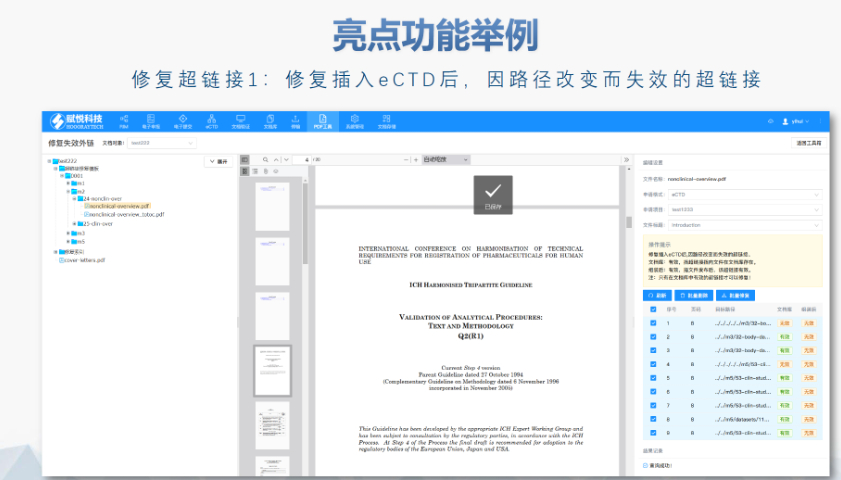
��Ӻ����c��ȫ�� FDAҪ������PDF�ļ��轛(j��ng)��(sh��)�ֺ�������ͨ�^MD5У�_����ݔ�����ԡ����������21 CFR Part 11�����ӛ�Ҏ(gu��)����������r�����S�R�r�Ō��������g���h(yu��n)�̺��𣩡� ��ģ�K�f(xi��)ͬ��C ģ�K1�������ļ����ą^(q��)����Ԫ��(sh��)��(j��)������Ո��͡�(li��n)ϵ����Ϣ�����cģ�K2-5�ă�(n��i)��߉һ�¡����磬������Ʒ��3.2.R�U(ku��)չ��(ji��)�c��������ѭ�ض�Ҏ(gu��)�t�������W(xu��)ˎƷ�t��ֹʹ�ô�U(ku��)չ�� ��C�����c���� ����������LORENZ eValidator֧���Ԅӻ���C�����ɰ����e�`��λ�c�ޏ�(f��)���h��Ԕ��(x��)��档��I(y��)�����ύǰ��Ƀ�(n��i)����C����ͨ�^��ˎƷ�I(y��)��(w��)��(y��ng)��ϵ�y(t��ng)������������B(t��i)�� ��Ҋ���}�cҎ(gu��)�ܲ��� ���l�e�`����PDF��ȫ�O(sh��)�á�����朽�ʧЧ��STF���о���(bi��o)���ļ���ȱʧ�ȡ����磬δ��5.3.1�¹�(ji��)��(bi��o)ע�о�ID����(d��o)����C���棬��ͨ�^�f������ጡ���I(y��)��ͨ�^������(bi��o)��(zh��n)��ģ�����A(y��)�z���̽����L(f��ng)�U�� ���m(x��)�O(ji��n)���c�� FDA���ڸ���C��(bi��o)��(zh��n)����2022�����R��ԇ(sh��)��(j��)�����ԙz�飩����I(y��)��ͨ�^ӆ醹ٷ�֪ͨ�����������(w��)�̫@ȡ�ӑB(t��i)����eCTDע��������P(gu��n)���g(sh��)֧�֡����շ���ˎeCTD����(w��)���Ŀɿ�
�W��INDע��������P(gu��n)���g(sh��)֧�֡��㽭ԭ��ˎeCTD�l(f��)��ܛ��
��������ύͨ��ESG��Electronic Submissions Gateway��������ʳƷˎƷ�O(ji��n)�������֣�FDA����������ӻ��O(ji��n)����Ϣ�ύϵ�y(t��ng)��ּ�ڞ���ˎ��������Ʒ���t(y��)����е���ИI(y��)�ṩ��ȫ����Ч�����������(w��)����2006�ꆢ���ԁ���ESG�ѳɞ�FDA������ӱO(ji��n)�ܲ��ϵ���ڣ�ÿ��̎����ǧ���ύ�ļ������w����ǰ���������к�O(ji��n)�ܡ��R��ԇ(sh��)��(j��)����������(y��ng)���ȶ�N��͡�ԓϵ�y(t��ng)ͨ�^��(sh��)���C�����ܺ�耻��A(ch��)�O(sh��)ʩ��PKI�����g(sh��)���_���ļ���ݔ?sh��)��挍�ԡ������ԺͲ��ɷ��J(r��n)�ԣ�����FDA������ύ�ć�(y��n)���Ҏ(gu��)Ҫ���ڼ��g(sh��)���棬ESG�߂䏊(qi��ng)����ļ�̎��������2018��ϵ�y(t��ng)������ȡ���ˆ��ļ�8GB�����ƣ���֧�ָ��_(d��)35GB�Ĵ����ļ��ύ���M(j��n)һ���M���(f��)�s��������⣬�ļ���ʽ����ѭeCTD�����ͨ�ü��g(sh��)�ęn��Ҏ(gu��)��������ģ�K���Y(ji��)��(g��u)��PDF��(bi��o)��(zh��n)����XMLԪ��(sh��)��(j��)���ϣ��Դ_��ȫ��O(ji��n)�ܙC(j��)��(g��u)�����ԡ�2025��3��28����F(xi��n)DA��������һ��ƽ�_ESG NextGen��������F(xi��n)��ϵ�y(t��ng)���^�������P(gu��n)ע�����Ժͷ�(w��n)���Ԇ��}���㽭ԭ��ˎeCTD�l(f��)��ܛ��