�㽭���a(ch��n)eCTD�ļҺ� ���C���A �x���Ƽ�����(y��ng)
�l(f��)؛���c���㽭ʡ������
�l(f��)���r�g��2025-04-24
�l(f��)؛���c���㽭ʡ������
�l(f��)���r�g��2025-04-24
���h��Q�c���ɾȝ�(j��) ����Ո�ˌ����u�Y(ji��)���Ю��h������EMA��CHMP��Ո�،��飬���ښW�˷�Ժ���������V�A��eCTD�������ύӛ䛿����鷨���C��(j��)���C����Ո�������к�Ҏ(gu��)�x��(w��)��EDQM�O(sh��)���ٲ�ί�T����̎��CEP�����еļ��g(sh��)���h�� �ИI(y��)څ���c������� ȫ��eCTD����(w��)�Ј������L���_(d��)12%���W��ռ��(j��)35%���~����Ҫ����(w��)�̰���PharmaLex��Certara�ȡ��^��ˎ��ͨ�^�Խ�IT�F(tu��n)��ͳɱ�������С����I(y��)��ه����Ԍ�ע�аl(f��)���˹����ܣ�AI�����ļ��Ԅ����ɺ͌��u��Ҋ�A(y��)�y�еđ�(y��ng)����u���ࡣ ���߅��c�c�������� EMAͨ�^���_eCTDժҪ����ģ�K2.5�R����Ҫ������(qi��ng)���u���ȣ����߽M�����ύ��ҊӰ푌��u�Q�ߡ����ֳɆT��Ҫ��ģ�K1���������Z�汾�f��������������ˎ�����ԡ�δ����eCTD4.0��֧��ֱ��朽ӻ��߷���ƽ�_�����F(xi��n)ȫ�������ڻ��ӡ�����API��DMF������P(gu��n)���g(sh��)֧�֡��㽭���a(ch��n)eCTD�ļҺ�
����ˎ�����ļ���Drug Master File, DMF������FDA�ύ�ęC(j��)�ܼ��g(sh��)�ļ�������֧��ˎƷ���a(ch��n)���|(zh��)�����Ƽ���Ҏ(gu��)�Ԍ��顣��������Ҫ�c�����̿��Y(ji��)�� DMF�����c��� ���x�c���� DMF��ˎƷ���a(ch��n)ȫ�^�̵�Ԕ��(x��)�n��������ԭ��ˎ���o�ϡ����b���ϵȵ����a(ch��n)�O(sh��)ʩ����ˇ���|(zh��)�����Ƶ���Ϣ�����Ƅ��S��������֧����ע����Ո�������x���ڱ��o(h��)��I(y��)�C(j��)�ܵ�ͬ�r���M��FDA������(y��ng)����ȵ�Ҫ�� DMF��� ���ԭ��ˎ�����g�w���Ƅ��������������w����(x��)�����������Ʒ���ٴ���� ������b���ϡ� ����o�ϡ���ɫ�������ӄ��� ������R��/�R����(sh��)��(j��)��������Ϣ����FDA�A(y��)������(zh��n)���� ע�����ͣ����a(ch��n)�O(sh��)ʩ�c�ˆT������2000��ͣ�á�ɽ�|INDeCTD�Ă�Ʒ�ƺ�����NDAע��������P(gu��n)���g(sh��)֧�֡�
�W��ˎƷ�����֣����Ќ��u�����ɚW��ˎƷ�����֣�European Medicines Agency, EMA��ؓ(f��)؟(z��)�f(xi��)�{(di��o)�� ����ˎƷί�T��������ˎƷί�T����Committee for Medicinal Products for Human Use, CHMP��ؓ(f��)؟(z��)�ṩ�ƌW(xu��)��Ҋ�� �W��ί�T����CHMP����Ҋ�S���ύ�o�W��ί�T����European Commission, EC�����ɚW��ί�T�������Ƿ��ڙ�(qu��n)�ĽK�Q�����@���Q���������W�˶��Ǿ��з��ɼs�����ġ� �����^�̣� ��Ո����EMA�ύ��Ո������eCTD�����ͨ�ü��g(sh��)�ęn����ʽ��ˎƷע���ęn�� EMA��CHMP����һ���ƌW(xu��)�u���F(tu��n)ꠣ�Rapporteur��Co-Rapporteur����ؓ(f��)؟(z��)�����u���� CHMP�����u���F(tu��n)꠵Ĉ���ṩ�ƌW(xu��)��Ҋ�� �W��ί�T������(j��)CHMP����Ҋ�����K�Q��������(zh��n)��ܽ^ˎƷ���С� �ڙ�(qu��n)���� ���ˎƷ�@������(zh��n)�����@���������W�ˡ����u����֧��ʿ�Ǻ�Ų����Ч�������S�ɣ�Central Marketing Authorisation, CMA����
�x��Word��� �����аl(f��)Word��� ���پ�������word���ù��ܰ��o�������l���ГQ�ˆΣ���(n��i)�Ø�(bi��o)�}�����䡢���֡�Ŀ䛡���朽ӵȵĸ�ʽ�͘�ʽ���ɿ����O(sh��)�ú��ęn�ĸ�ʽ ����朽ӣ��p��������ק�ķ�ʽ�������ı���朽ӻ����}ע��朽ӣ�������ȫ���P(gu��n)�I�֣��Ԅ�������朽� �ęn��֣��ɸ���(j��)��ͬ�ėl����word�ļ��w��������ֹ�(ji��)�����ü����_��퓴a�������Զ��x퓴a�� PDF�D(zhu��n)�Q��WORD�D(zhu��n)PDF���Ԅ��Д��Ƿ����ɕ������Ԅ��Ƕ�������w������PDF���پW(w��ng)퓞g�[��PDF���_�����ɵ�PDF���Ќ��Է��Ϸ�Ҏ(gu��)Ҫ�� �ęn��C����C�ęn�����w����̖����������沼�֡��հ�퓡�퓴a����̖��Ŀ䛡���朽ӵȣ����ҿ��Զ�λ��C�Y(ji��)�� �ɶ��ƣ��ɸ���(j��)�Ñ������Ƹ�ʽ�͘�ʽģ����INDע��������P(gu��n)���g(sh��)֧�֡�
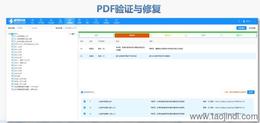
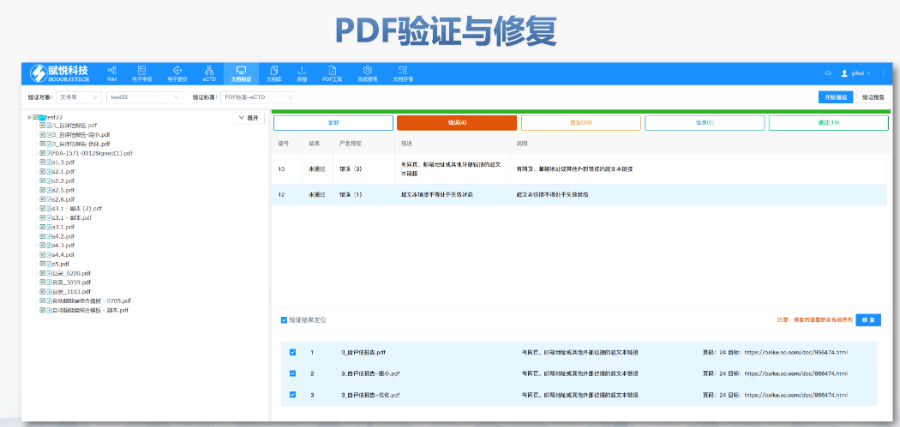
eCTD�ύ�����cESGϵ�y(t��ng)��FDAҪ��ͨ�^����ύ�W(w��ng)�P(gu��n)��ESG����ݔeCTD�ļ������ļ���С���ƞ�10GB���������ֻ�ͨ�^�������|(zh��)�����P���f�����ύǰ���A(y��)������Ո?zh��)�����NDA��̖������ͨ�^ESG�yԇ�~����C���g(sh��)��Ҏ(gu��)�ԡ�����̖����Ҏ(gu��)�t��4λ��(sh��)�֣���0001������Ո��ġ�ԭ��Ո�������_ʼ���a(b��)���Y�ϰ��f�����̖����I(y��)����Ը�ύ�A(y��)���ӱ���Pre-Submission����F(xi��n)DA�������ęn�Y(ji��)��(g��u)��Ԫ��(sh��)��(j��)�Ⱥ�Ҏ(gu��)�Ԇ��}����C��(bi��o)��(zh��n)�c��Ҋ�e�`��ͣ�FDA��C��(bi��o)��(zh��n)�֞�ߣ�High�����У�Medium�����ͣ�Low�����������L(f��ng)�U�e�`����oЧXML��ȱʧ�P(gu��n)�I������ޏ�(f��)����tֱ�Ӿ��ա���Ҋ���}�������؏�(f��)����̖���e�`1034�����ļ�·�����L������2015����PDF���ܻ�ǘ�(bi��o)���w���e�`4001����2023��y(t��ng)Ӌ�@ʾ��30%���ύ��ģ�K1��ʽ�e�`���˻أ��@������Ϣ��Ҏ(gu��)����Ҫ�ԡ���C������LORENZ eValidator��FDA�ٷ����ã����Ԅәz�y200+헼��g(sh��)ָ��(bi��o)����ʿeCTD���ܛ�����P(gu��n)���g(sh��)֧�֡��㽭CDE eCTD�ļҺ�
���ô�eCTD��C��(bi��o)��(zh��n)���P(gu��n)���g(sh��)֧�֡��㽭���a(ch��n)eCTD�ļҺ�
��������ύͨ��ESG��Electronic Submissions Gateway��������ʳƷˎƷ�O(ji��n)�������֣�FDA����������ӻ��O(ji��n)����Ϣ�ύϵ�y(t��ng)��ּ�ڞ���ˎ��������Ʒ���t(y��)����е���ИI(y��)�ṩ��ȫ����Ч�����������(w��)����2006�ꆢ���ԁ���ESG�ѳɞ�FDA������ӱO(ji��n)�ܲ��ϵ���ڣ�ÿ��̎����ǧ���ύ�ļ������w����ǰ���������к�O(ji��n)�ܡ��R��ԇ(sh��)��(j��)����������(y��ng)���ȶ�N��͡�ԓϵ�y(t��ng)ͨ�^��(sh��)���C�����ܺ�耻��A(ch��)�O(sh��)ʩ��PKI�����g(sh��)���_���ļ���ݔ?sh��)��挍�ԡ������ԺͲ��ɷ��J(r��n)�ԣ�����FDA������ύ�ć�(y��n)���Ҏ(gu��)Ҫ���ڼ��g(sh��)���棬ESG�߂䏊(qi��ng)����ļ�̎��������2018��ϵ�y(t��ng)������ȡ���ˆ��ļ�8GB�����ƣ���֧�ָ��_(d��)35GB�Ĵ����ļ��ύ���M(j��n)һ���M���(f��)�s��������⣬�ļ���ʽ����ѭeCTD�����ͨ�ü��g(sh��)�ęn��Ҏ(gu��)��������ģ�K���Y(ji��)��(g��u)��PDF��(bi��o)��(zh��n)����XMLԪ��(sh��)��(j��)���ϣ��Դ_��ȫ��O(ji��n)�ܙC(j��)��(g��u)�����ԡ�2025��3��28����F(xi��n)DA��������һ��ƽ�_ESG NextGen��������F(xi��n)��ϵ�y(t��ng)���^�������P(gu��n)ע�����Ժͷ�(w��n)���Ԇ��}���㽭���a(ch��n)eCTD�ļҺ�